Today, software is an integral aspect of the medical and healthcare industry. The accelerated changes brought about by technology have created a requirement for various types of solutions which help the healthcare providers to engage with patients effectively and quickly. Such software solutions have also helped to maintain market relevance.
SAMD or SaMD, abbreviated for software as a medical device, is one such novice and revolutionary concept of the software solutions in the medical and healthcare industry. The implementation of Software as a Medical Device is becoming increasingly popular and can be applied across a broad range of technological platforms.
In reference to medical devices, SAMD is only one of three types of softwares. The other two are software in a medical device software which is a type of software that is integral to a medical device and software that is implemented for either manufacturing or maintaining a medical device. In this article, we concentrate on SAMD or software as a medical device to understand its integration and impact upon the medical devices and the healthcare industry.
Software as a medical device is specifically intended for medical purposes. It’s a type of software that’s functional by itself and can function without the hardware.
It can be applied to support and evolve various applications. SAMD not only improves efficiency and accuracy in healthcare solutions but also provides alternative options for treatment. While it’s extremely advantageous in advancing healthcare solutions, it does pose a certain set of challenges such as data privacy and cybersecurity concerns.
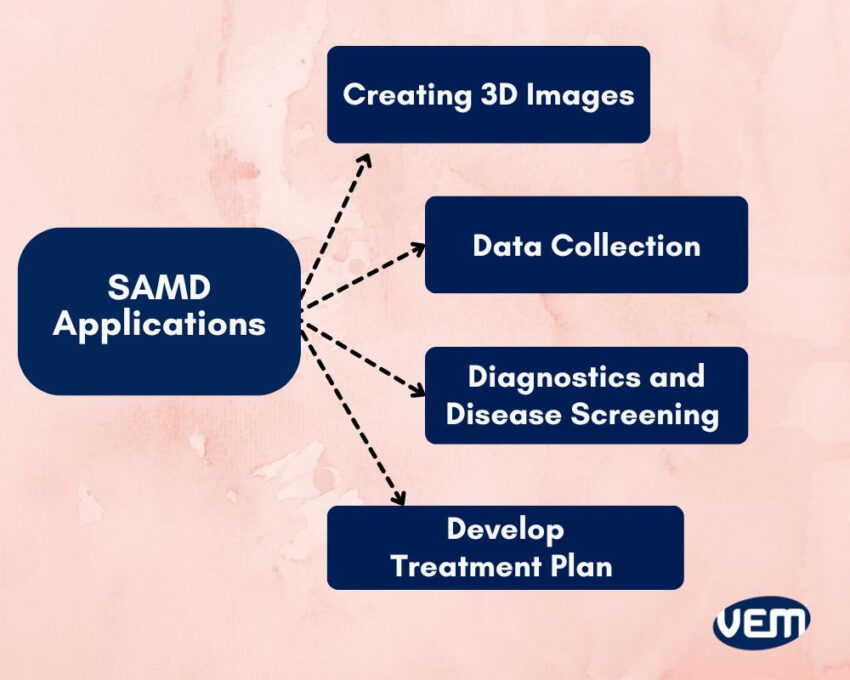
One of the significant applications of Software as a medical device in the healthcare industry is when, the SAMD creates 3D images to help surgeons and physicians visualize better. SAMD makes it possible to create 3D images of patient’s medical images.
SAMD can collect and process a large amount of data from various sources such as X-rays, MRI scans, etc. This data can be further used to create 3D models which can be used for either diagnosis and developing treatments.
It can also collect patient data in real-time and from multiple sources thereby, enabling medical professionals to view and analyze data from a remote location.
It enables healthcare professionals to view and analyze images for diagnostic purposes. SAMD can be developed to view images from an MRI, X-ray or ultrasound which can be further analyzed for diagnostic purposes. You should note that SAMD collects data from other devices, but analyzes the data by itself.
SAMD collects and analyzes data that can be used to develop treatment plans by the physician or the surgeon. In addition, it’s algorithm can develop a treatment plan for a specific condition which can be monitored by the medical professional and further, implemented.
It is imperative to understand whether the software in itself can be characterized as a medical device or not. If the product meets the characterization of a medical device, then the following definitions from the IMDRF and FDA must be delved upon:
SiMD is a type of an embedded software within a medical device that is used to either power, run or operate the device. SiMD, abbreviated for software in a medical device, can function by either powering the mechanics of the medical device or creating a graphical interface for the device. For e.g. An insulin pump in which the software controls the insulin delivery for the patient.
On the other hand, SAMD must be able to perform all its function with or without the hardware of the medical device for it to be categorized as a software in a medical device.
SAMD is a medical device, and is thus regulated as such. Just like any other medical device, it requires a QMS, abbreviated for quality management system.
If the product is to be marketed in the United States, then the FDA 21 CFR Part 820 regulation that outlines the requirements of Quality System Regulations (QSR) needs to be established and followed. For the European region, the EU 2017/745 i.e. EU MDR needs to be followed. You can read more about the Medical Device QMS here.
Just like how, the US and the European region have different risk categories for medical devices, the IMDRF and IEC 62304 have different methods of categorizing SAMD. In this section, we delve into the various categories and their correlation with each other.

The US FDA system classifies software as a medical device through the same risk classes as it does for other medical devices i.e. Class I, Class II, and Class III. In the European region, the medical device software risk class and “Rule 11” is applied. You should note that, there is no MDSW-specific risk classification in the US as it is in the European region. The medical device software also utilizes the same risk classification as it does with other medical devices i.e. class I, IIa, IIb, and III. A major distinction between the US and EU classification system is that the EU MDR outlines in Rule 11 about how to determine your medical device software risk class.
The IMDRF categorization is not extensively employed but when the SAMD needs to be marketed in the EU region, the IMDRF categorization can be particularly helpful to determine the SAMD’s risk class.
IMDRF categorizes SAMD into 4 risk categories which are a combination of 2 factors and divided into three levels. The following infographic depicts the IMDRF risk categorization for Software as a medical device:
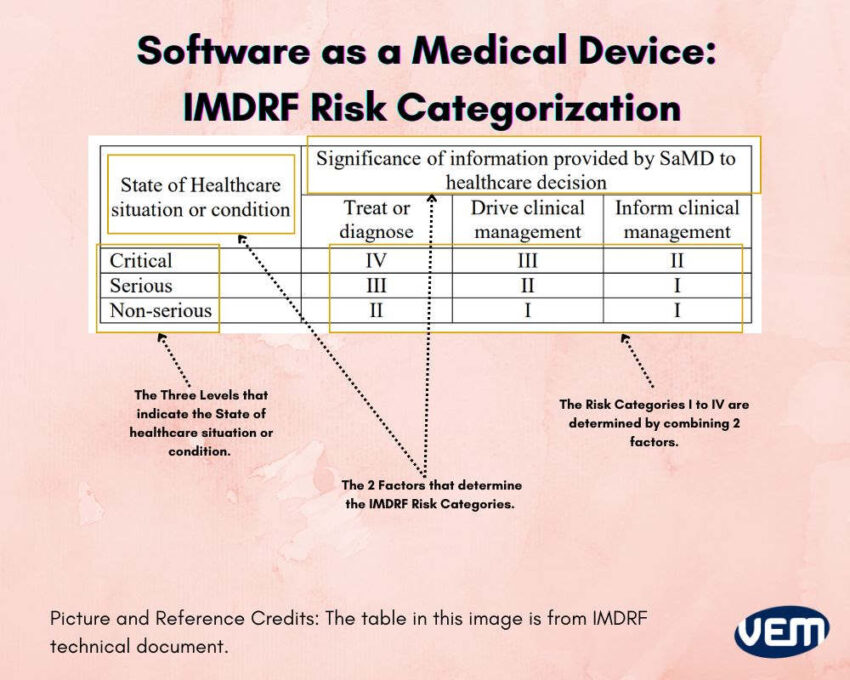
The three levels in IMRDF risk categorization indicate the following State of Healthcare situation or condition:
The 4 risk categories i.e. I to IV are a combination of 2 factors. Let’s understand these categories further:
You can read more about the IMDRF risk categorization in it’s technical document here.
IEC 62304 is an international standard for software safety classification. It was last reviewed and confirmed in 2021 and you can read the technical document here. This system classifies based on the injury severity that can be caused in case of software failure. The software safety classification as per IEC 62304 is depicted in the below infographic:
FMEA increases medical devices’ accuracy, sensitivity, and reliability through its thorough analysis. It proactively identifies the possibility of malfunction issues in medical devices during the design and development stages thereby, increasing patient care.
It improves design for manufacturing and assembly (DFM/A) and enables collaboration between design and operations. In addition, FMEA continually improves manufacturing and post-marketing processes directly or indirectly.

You should note that IEC’s 62304 software safety classification does not indicate that the software is safe to use or not. The safety of SAMD is largely dependent upon the design and development process.
SAMD is subject to the same requirement by the various regulatory bodies as other medical devices. Thus, all of the postmarket requirements of FDA’s QSR, EU MDR or IVDR will be applied to fulfill the past-marketing requirements for a SAMD or Software as a medical device.
If a significant change is required in the SAMD, you may need to resubmit the documentation. In this section, let’s understand how you can implement SAMD document changes for resubmission of the device in the market:
Just like other medical devices, SAMD can be updated and resubmitted in the US market via notifying FDA of the change. Either a new 510(K), a special 510(k), or a pre-market approval supplement can be submitted to facilitate SAMD changes in the US.
FDA has also released a guidance document in 2017 for manufacturers to understand
and facilitate the process of resubmission. You can read Deciding When to Submit a 510(k) for a Software Change to an Existing Device here.
The requirements to implement changes for SAMD in the European region are similar to the FDA. You can read EU MDR Annex X to further understand the requirements for implementing changes in your device.
Artificial intelligence is one of the most novice technologies that is ever evolving and being applied to medical devices. It includes incorporating ML, abbreviated for machine learning that improves or alters its outputs as it gains more information through an increasing amount of inputs.
While this technology takes an incredible stride in producing accurate results for the medical device industry, it does cause changes in the algorithm to improve it. To address the same, and understand if enough information has been gathered for a new submission, FDA has released a Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD). In this particular proposed framework, the AI/ML modifications are proposed to be based upon a “predetermined change control plan” which basically establishes parameters for anticipated modifications. The framework is evolving but the principles outlined here are particularly useful for manufacturing SAMD with AI/ML technologies.
IEC 62304 is a standard requirement of the process standard which indicates an optimized procedure for structuring processes to create safe and effective software. As per IEC 62304, the following 5 processes need to be adhered to during the software lifecycle:
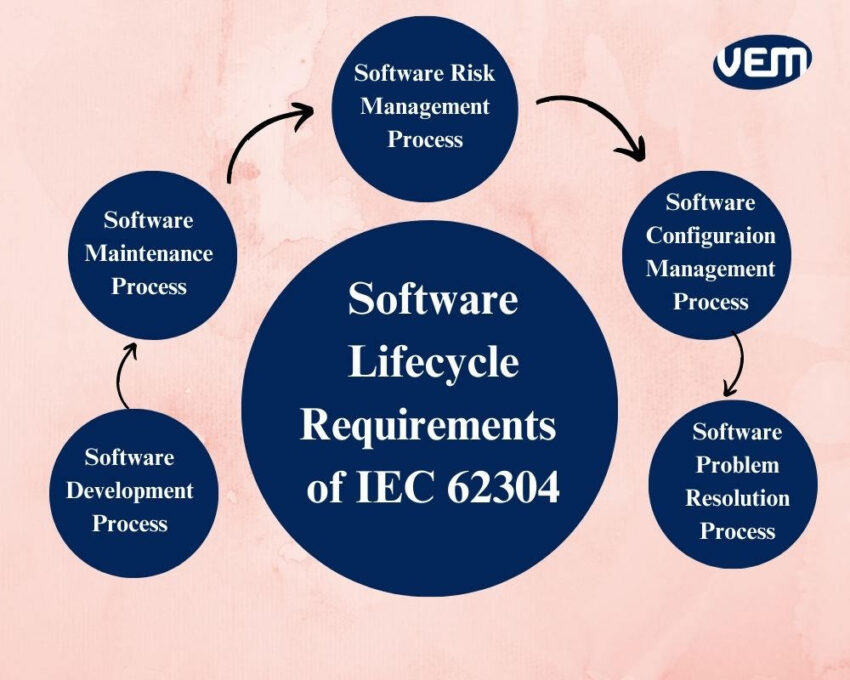
This process plans the development process that begins with the planning and ends with the release of the software. It includes outlining requirements analysis, architectural design, software unit implementation and verification, the integration of software, testing of the integration and system.
The next step in the IEC 62304 is the software maintenance process. This stage includes outlining the maintenance plan, analyzing and implementing modifications.
The requirements of risk management in IEC 62304 are similar to those outlined in ISO 14971 which is an international standard for the application of risk management to medical devices. Just like other medical devices, software in a medical device is also expected to follow similar requirements of not only IEC 62304 but also ISO 14971.
Software configuration tracks and records the minutest details for your software. IEC 62304 indicates that details such as change control, identification and status accounting of configuration are recorded in this process.
Typically, there are various types of problems that are encountered throughout the lifecycle of the software. This stage outlines the various requirements for a problem resolution process. It includes investigating the problems, creating the reports and maintaining records. It also involves using change control processes, verifying problem resolutions and validating the development of SAMD.
It is crucial to consider cybersecurity when we discuss software development for or in the medical device industry.
Cybersecurity is a relatively novice segment that’s still evolving in the medical device industry but you should note that in the past decade, there have been various attacks upon the healthcare industry. It’s thus best to address and involve cybersecurity throughout the design process!
You can read more about FDA’s updates on Cybersecurity here and the guidance document by IMDRF here.
Developing and incorporating a SAMD in the market is a complex process. It’s crucial to understand the various requirements of the regulatory bodies.
VEM Medical has over 20 years of experience in manufacturing high-quality medical devices. We can help you with an end-to-end solution for developing a SAMD and more.