Medical device classification helps to identify and label crucial medical devices into diagnosis, prevention, monitoring, and treatment segments of the healthcare industry. They can vary from being simple and posing little or no risk to the patient or user to being extremely complex and life-sustaining.
It is imperative that medical device manufacturers understand the various medical device classifications and familiarize themselves with the applicable regulations of their intended markets. You should note that different countries have different requirements.
For instance: the regulatory bodies in the United States, Canada, and Europe differ in their classification and requirements however; there are various overlaps between these international medical device regulatory bodies. Understanding the medical device classification helps to launch innovative devices and products into its intended market. In this article, we discuss the medical device classification by some of the most popular regulatory agencies in detail.
What is Medical Device Classification?
There is a wide variety of medical devices available and they are regulated on a risk-based classification system. You should note that the class of the medical device is directly proportional to the risk of the device i.e. If the risk is higher, then the class of the medical device will be higher. The higher the classification of the medical device, the more stringent and complex the data requirements are and the sole purpose is to ensure that the medical device is effective and safe to use for its intended purpose.
Purpose of Medical Device Classification
The classification of medical devices is not only for following standards set by regulatory agencies but they also help manufacturers to develop the device. Let’s understand the significance of medical device classification:
- Medical device classification enables manufacturers to determine their action steps before entering the market to sell the product.
- Medical device classification helps to establish various requirements during the product development phase. This is especially the case with design controls.
- It helps to determine the cost of introducing the device into the market.
- It further estimates the time that will be taken to develop the medical device.
Medical Device Classes Defined By Regulatory Agencies
Medical device classes are classified and defined by various regulatory agencies. Each regulatory agency has defined several different medical device classes. Typically, the medical device classification is dictated by the perceived risk of the product. The following agencies are some of the most prominent regulatory agencies when it comes to medical device classification:
- European Commission
- U.S. Food & Drug Administration
- Health Canada
In Canada and the European Region, medical devices are grouped into four classes whereas in the United States, they are grouped into three classes.
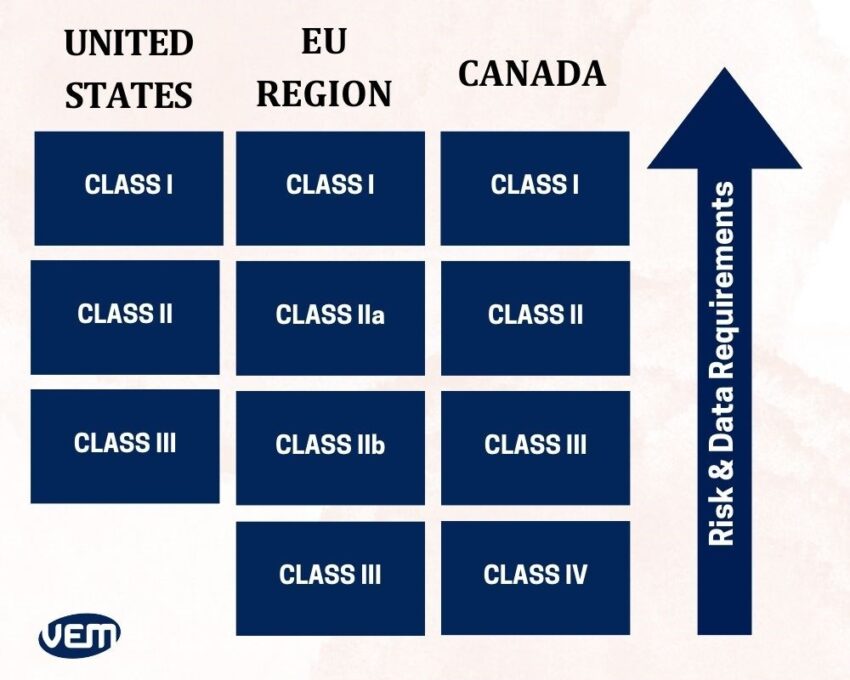
FDA Medical Device Classification
Medical devices are regulated by the FDA, abbreviated for Food & Drug Administration in the United States. The specific sector responsible within the FDA is the Center for Devices & Radiological Health (CDRH) and its primary goal is to protect and promote public health by ensuring medical devices are safe for their intended use.
FDA’s approach
The FDA medical device classification is primarily based on the risk that the medical device poses. The medical devices are classified as Class I, Class II, and Class III by the FDA in the United States.
- Class I indicates medical devices that require general control and are recognized as ‘low to moderate risk’. They are denoted as Class I devices by the FDA. It is not required to submit a premarket notification for most of the Class I devices.
- Class II encompasses medical devices that require general or special control such as performance standards, postmarket surveillance, and premarket notification requirements (510(k) submission). They are recognized as ‘moderate to high risk and are denoted as class II devices by the FDA.
- Class III encompasses medical devices that require general and special controls as well as pre-market approval (PMA). They are recognized as the ‘highest risk’ of all the classes and denoted as class III devices by the FDA.
How can you identify the correct classification for your medical device?
To correctly identify the medical device classes, one must keenly note the distinction between intended use and indications for use. Let’s understand these terms further:
- Intended Use: This term describes the general purpose or function of the medical device.
- Indications for Use: This term describes how the device is used to diagnose, treat, or prevent a disease or a condition of the target patient population.
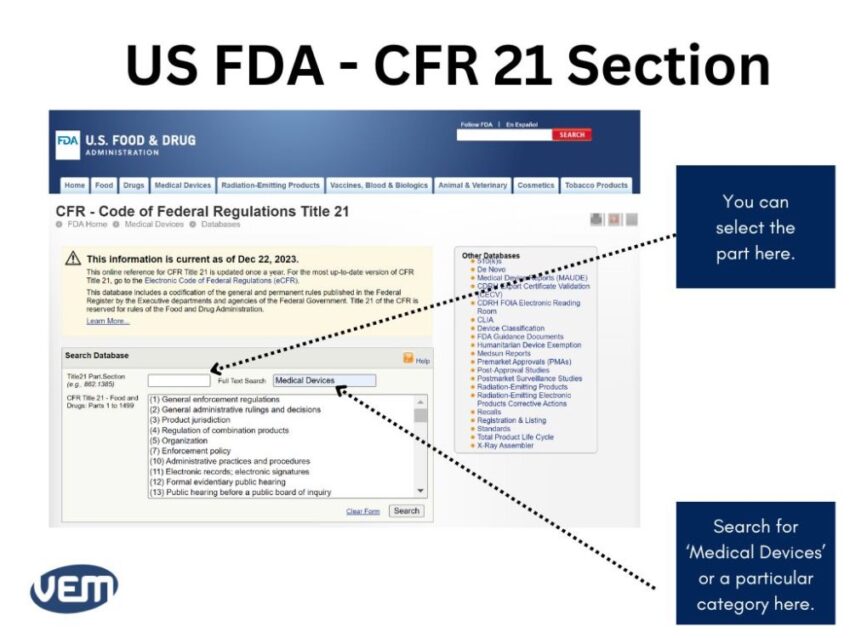
Once you define intended use and indications for use for your medical device, you can locate the product codes and the possible regulations. FDA has established several categories in CFR Title 21 – Food and Drugs. You can search for your category of medical devices here.
The following image depicts the current US FDA CFR 21 section:
Case-study for Identifying Information upon a Particular Device
Let’s consider that you need to locate information for a caries detection dental device. In such a case, the information for your medical device will be available in Part 872 dental devices. You can view the CFR Code by scrolling below:
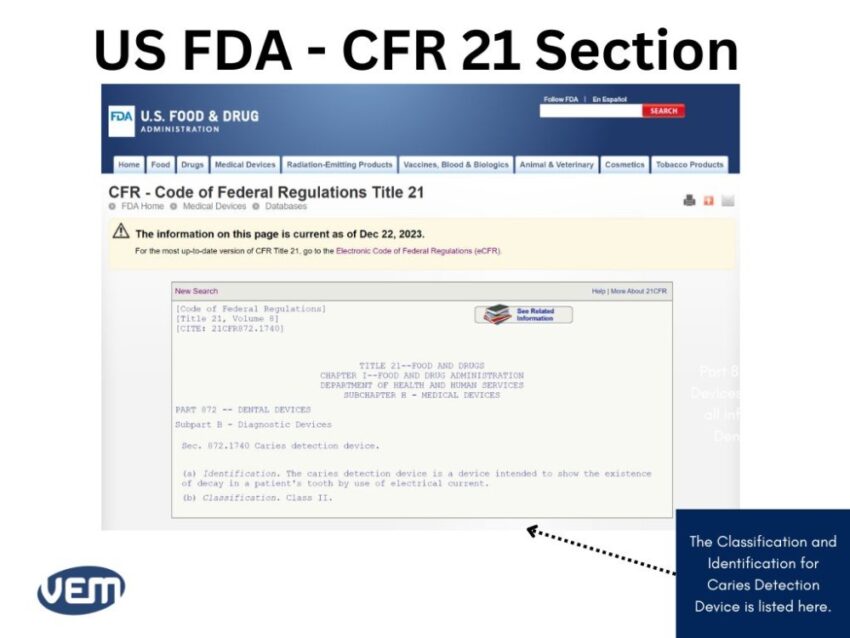
The CFR Title Codes for Medical Devices are between Part: 800 to 1299. Once you find the CFR code that could be the likely fit, you can click on it for further details to get the following page:
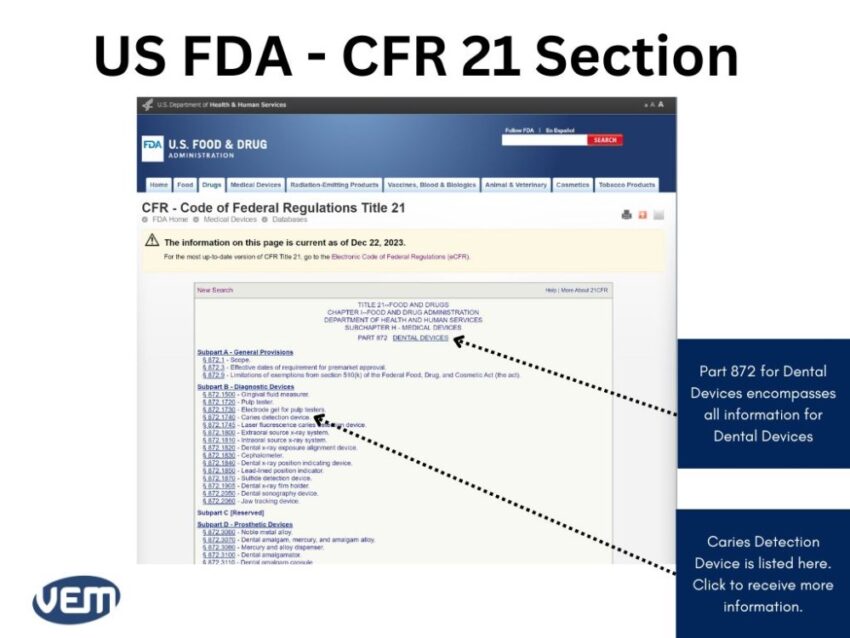
In this case, the information is listed in the 872.1740 section. You can now view the information on caries detection devices by clicking here to get the following page:
The details enlisted here indicate that the product is a Class II medical device.
Determination of Medical Devices in the U.S. Market
Once you understand the category, product codes, and applicable regulations, you can determine the exact pathway to register your product with the FDA.
If only general controls apply, then the medical device is exempt; thus, no formal FDA submission is required. You should note that it is required to register your establishment with the FDA and list the product.
On the other hand, if your medical device requires special controls, then a 510(k) clearance must be submitted to the FDA to enter the market after which, you need to register your establishment and list the product.
You should note that if the medical device requires premarket approval, the FDA PMA process must be followed to receive approval before going to market.
European Medical Device Regulation – EU MDR Classification
Medical device regulations in the European Union (EU) are established through EU MDR 2017/745. This is regulated by the European Commission (EC) and is applicable in the European Union from 26 May 2021.
EU MDR Classification for Medical Devices

You should note that the European Union has a similar medical device classification system as the FDA however; all medical devices that are marketed in the European Union must first obtain a CE marking. Let’s understand the 4 categories of EU MDR Classification:
Class I Devices
Class 1 medical devices include your everyday devices that are low-risk and non-invasive. Examples of class 1 devices in the European Region are bandages, compression hosiery, and walking aids.
Class 1 devices do not require a conformity assessment test by a notified body however; a complete technical file is mandatory for such devices. You can read more about the Fact Sheet for Class 1 Devices here.
Class IIa Devices
Class IIa medical devices are typically installed within the body for the short term i.e. only between 60 minutes and 30 days. They constitute a low to medium risk. Examples of class IIa devices in the European Region are hearing aids, blood transfusion tubes, and catheters.
Class IIa Devices require both, a technical file to be mandatorily completed by the manufacturer and a conformity assessment test carried out by a notified European regulatory body.
Class IIb Devices
Class IIb medical devices are more complex than class IIa devices and are installed in the body for 30 days or longer. They are generally medium to high risk. Examples of Class IIb devices are ventilators and intensive care monitoring equipment.
Its requirements are the same as Class IIa devices that require both a technical file and a conformity assessment test however; there is an added requirement of a device-type examination by a European notified body.
Class III Devices
Class III medical devices are high-risk devices such as balloon catheters and pacemakers. Class III device approval requires a complete quality assurance system audit. In addition, the device’s design and the device itself will undergo a thorough examination by a European Notified Body.
CE Marking
It is imperative to obtain a CE marking to market in Europe. This process requires extensive technical documentation and the requirements to obtain CE-marking are based on the European System Commission.
CE Marking denotes that the products sold in the EEA (European Economic Area (EEA)) have been assessed thoroughly to meet the various requirements of safety, health, and environmental protection. You can read about CE marking here.
EU MDR Device Categories
Certain rules apply to each of the broad categories. Let’s understand these categories further:
- Non-Invasive: This category includes any device that does not penetrate the body either through the surface or an orifice. They are typically Class I devices; however, in some cases, they could be Class II devices or higher as well.
- Invasive: This category encompasses any device that penetrates the body, either through the surface or an orifice. Even if a part of the device is invasive, it will be categorized as invasive.
- Active: An active device is any type of device that depends on an energy source to operate. This energy source can be generated either by the human body or by gravity.
The rules for each of these categories are outlined in Annex XVI of the medical device regulation.
Canada Medical Device Classification
Medical Device Classifications in Canada are established and regulated by Health Canada. You can read more about the Medical Device Classification in Canada in detail here.
Health Canada requires medical device manufacturers to follow a guidance document on the risk-based classification system for non-in vitro diagnostic devices. This is mandatory to be implemented for medical device manufacturers to market and sell products in Canada.
Classification Groups by Health Canada
In Canada, medical devices are categorized based on the risk associated with their use. Health Canada defines the following four groups of non-in vitro diagnostic medical devices:
- Class I medical devices – These devices are non-invasive and they are of the lowest risk.
- Class II medical devices – Class II devices are invasive and they are typically low-risk devices.
- Class III medical devices – Class III devices are active devices and they are of moderate risk.
- Class IV medical devices – These devices are classified under special rules and are of the highest risk.
The medical devices of Class I do not require a license in Canada but the medical devices of Class II, III, and IV must be licensed before they are sold or imported in Canada.
Medical Device Single Audit Program (MDSAP) and ISO 13485 Certification
There are a set of rules that are applied to each of the broad categories and they should be followed to determine the risk classification of the device.
If a medical device is categorized as Class II or higher, it must undergo the Medical Device Single Audit Program (MDSAP) and obtain an ISO 13485 certification.
- To gain MDSAP certification, medical device manufacturers must complete the application and review, Off-site documentation audit, and On-site audit.
- ISO 13485 are standards set for QMS requirements that are to be implemented for the design and manufacturing of medical devices. You can read about the ISO 13485 standard here.
You should note that the MDSAP compliance is based on meeting the guidelines from the ISO 13485 standard on QMS.
Manufacturers when selling medical devices in Canada, need to undergo annual reviews. In addition, they will need to undergo a recertification audit every third year.
Contact Us
Medical device classification is integral to understanding the key regulatory requirements for the manufacturing of medical devices. At VEM-Tooling, we have a team of design experts and engineers who have a vast experience of over 20 years in manufacturing high-quality medical device plastic parts
If you are looking for a medical device solution, you can contact us and talk to our experts.


